
Research Article
A systems pharmacology approach for targeted study of potential inflammatory pathways and their genes in atherosclerosis
Sanaa Almowallad 1, Raja Jeet 2 and Mohammad Mobashir 3,*
1Â Â Â Department of Biochemistry, Faculty of Sciences, University of Tabuk, Tabuk, 71491, Saudi Arabia; salmowaled@ut.edu.sa (S.A.).
2Â Â Â G D College, Begusarai, 851101, Bihar, India; rajajeet10@gmail.com (R.J.).
3Â Â Â Department of Biomedical Laboratory Science, NTNU, NO-7491 Trondheim, Norway.
*Â Â Â Correspondence: mohammad.mobashir@ntnu.no (M.M.)
| Citation: Almowallad S, Jeet R, and Mobashir M. Â A systems pharmacology approach for targeted study of potential inflammatory pathways and their genes in atherosclerosis. Glob. Jour. Bas. Sci. 2024, 1(2). 1-12.
Received: September 21, 2024 Revised: October 15, 2024 Accepted: October 29, 2024 Published: November 09, 2024 doi: 10.63454/jbs20000006 ISSN: 3049-3315 Download PDFÂ file |
 Abstract: Atherosclerosis is a widespread and chronic progressive arterial disease that has been regarded as one of the major causes of death worldwide. It is caused by the deposition of cholesterol, fats, and other substances in the tunica intima which leads to narrowing of the blood vessels, loss of elasticity, and arterial wall thickening, thus causing difficulty in blood flow. For a long time, one of the most crucial approaches to the treatment and prevention of cardiovascular illnesses has been the use of natural products. Numerous studies on natural chemicals that are beneficial against atherosclerosis have been carried out in recent decades due to the growing interest in natural products, including medicinal herbs. The primary cause of coronary heart disease, a chronic inflammatory condition marked by fat buildup in the artery wall, is atherosclerosis. Since inflammation is its precursor, seaweed-derived polysaccharides that target inflammatory genes are a viable source of anti-inflammatory drugs. After preparing the list of herbal medications and predicting the suspected indicators for atherosclerosis, we looked for possible binding targets. The binding model of fucoidan and alginate with targeting genes was predicted using computational methods such as molecular docking and Swiss-ADME. Molecular docking experiments showed that Fucoidan could block genes involved in inflammation. Furthermore, Fucoidan’s sulphate group gives it unique properties in the GI tract and molecular dynamics. These findings suggest that sulphate polysaccharides may have anti-atherogenic properties. We used the publicly available dataset to investigate the top-ranked genes based on network-level understanding of gene expression patterns and their effects on functions. The pathways that were most affected were phagosomes, cell adhesion molecules, haematopoietic cell lineage, cytokine-cytokine receptor interaction, osteoclast differentiation, antigen processing and presentation, and critical immune signalling pathways. Additionally, based on networks of DEGs, the genes that were most connected were PTPRC, ITGB2, HCLS1, RAC2, LAPTM5, CD37, CD53, CD48, SYK, HCK, TYR0BP, FERMT3, COR01A, LCP1, and phagosome. Here, we have investigated the possible connections between the marine medications Fucoidan and Alginate-N, as well as their possible targets in atherosclerosis.
Keywords: Natural drugs (NDs)/Herbal drugs (HDs); Fucoidan and Alginate-n; atherosclerosis; metabolic disorders; genes and pathways; biological networks
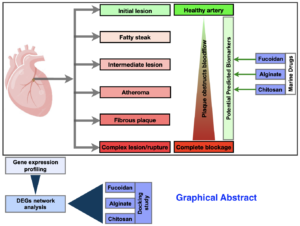
1. Introduction
One of the main causes of death and morbidity worldwide is atherosclerosis. Peripheral vascular disease, myocardial ischaemia, heart failure, heart attacks, and strokes are all caused by it. Inflammation, damage, and malfunction of endothelial cells in the heart are hallmarks of atherosclerosis. Acute cardiovascular disease is brought on by unstable atherosclerotic plaque rupture, arterial stenosis, or occlusion brought on by platelet aggregation and thrombosis. Atherosclerosis is a chronic inflammatory disease[1-6]. Proinflammatory cytokines, inflammatory signalling pathways, adhesion molecules, and bioactive lipids all contribute to the inflammation associated with atherosclerosis. The impact of inflammation and the systemic inflammatory signalling pathway on atherosclerosis, the part related signalling pathways play in inflammation, the development of atherosclerosis plaques, and the potential for atherosclerosis treatment through inflammation inhibition are all covered in this review. Plaque accumulation in the wounded area, arterial constriction, cholesterol deposition on the arterial wall, and monocyte adhesion to the endothelium are all consequences of endothelial injury. This mechanism causes chronic inflammation, which ultimately results in thrombosis or stenosis. The lipid-driven, multifocal, smouldering immuno-inflammatory illness known as atherosclerosis affects the medium and large arteries. Leukocytes, intimal smooth muscle cells, and endothelial cells are the primary players in the development of this disease. The most severe consequences of atherosclerosis, including heart attacks and strokes, are brought on by superimposed thrombosis. Therefore, the important topic is not why atherosclerosis starts, but rather why, after years of slow progression, atherosclerosis becomes complicated by luminal thrombosis. If thrombosis-prone plaques could be identified and prevented, atherosclerosis would be a far less dangerous condition. The vast majority of deadly coronary thrombi are caused by plaque rupture. Compared to women, coronary thrombosis is more frequently caused by plaque rupture in men[3, 4, 7-18].
As blood pressure, smoking, and low-density lipoprotein (LDL) cholesterol levels have dropped, the risk factor profile has changed. The preventive properties of high-density lipoprotein have been questioned by recent research, which now focusses on triglyceride-rich lipoproteins as well as low-density lipoproteins as the causes of atherosclerosis[9-11, 16, 19-21]. Atherosclerosis’s non-traditional causes, like sleep disturbances, sedentary lifestyles, the microbiota, air pollution, and environmental stress, have also drawn more attention. Leukocytes and inflammatory pathways connect both established and new risk factors to the changed behaviour of arterial wall cells. Investigating the pathophysiology of atherosclerosis has brought attention to the bone marrow’s role: clonal haemopoiesis, a previously unknown but frequent and significant age-related contributor to the risk of cardiovascular disease, can result from somatic mutations in stem cells. The idea of “vulnerable plaque” has given way to more detailed descriptions of the mechanisms behind the thrombotic consequences of atherosclerosis. These developments in our knowledge of the biology of atherosclerosis have made it possible to develop therapeutic approaches that could enhance the prevention and management of the now common atherosclerotic conditions. Ruptured plaques exhibit angiogenesis, adventitial inflammation, and outward remodelling. They consist of a big, lipid-rich core, angiogenesis, adventitial inflammation, outward remodelling, and a thin, fibrous cap with many macrophages and few smooth muscle cells. Plaque rupture is the most common cause of coronary thrombosis. Distinct patho-anatomical features of ruptured plaques, and consequently, rupture-prone plaques, may help with in vivo image recognition[8, 10, 12, 13, 16, 18, 19, 22, 23].
Atherosclerosis is by far the most prevalent underlying cause of peripheral arterial disease, carotid artery disease, and coronary artery disease. Thrombosis, which happens when a ruptured or eroded atherosclerotic plaque is superimposed on another ruptured or eroded atherosclerotic plaque, is what causes life-threatening clinical events including acute coronary syndromes and stroke. Atherosclerosis by itself is rarely fatal. Therefore, the key question is not why atherosclerosis develops, but rather why none or a small number of plaques out of many in a particular person seem to experience a thrombosis-prone and fatal phase over their lifetime. Atherosclerosis, a chronic inflammatory blood artery disease, is the primary cause of cardiovascular disease, a major cause of death globally. Along with cholesterol that enters from the circulation, atherosclerotic lesions also contain macrophages, T lymphocytes, and other immune response cells. While interfering with regulatory immunity speeds up disease, targeted ablation of genes expressing proinflammatory cytokines and costimulatory factors reduces disease in mice models. Atherosclerosis has been linked to both innate and adaptive immunological responses, with elements of low-density lipoprotein that carry cholesterol causing inflammation, T cell activation, and antibody formation as the illness progresses[3, 5, 8, 12, 13, 16, 17, 19, 21, 24-29].
Since the beginning of civilisation, natural goods have been prized for a number of uses, including medicinal ones. As they have been used to treat numerous other conditions, medicinal herbs have also been used to cure atherosclerosis. The mechanisms of action of several herbs used to prevent atherosclerosis have only lately been clarified by the use of cellular models for anti-atherogenic natural product screening or in-depth research on certain plants. To better comprehend these plants, recent studies on natural products—particularly medicinal herbs—were categorised and described based on their mechanisms of action (MOAs) against atherosclerosis.
- Methods
In order to find possible targets and herbal medications that can interfere with the activated signalling pathways, we proposed to examine the activated signalling pathways of individual atherosclerosis. In particular, we use an integrated method to first discover the genes that are up- or down-regulated in patients with atherosclerosis, and then we identify the components of the pathway. Lastly, we mapped the upstream/downstream signalling for the medicines alginate and fucoidan.
The project’s objective was to identify genes with altered expression in macrophages from atherosclerosis patients compared to macrophages from healthy individuals. To that end, we first selected the data we were interested in (raw expression datasets) GSE9874 (total 60 samples, 30 normal and 30 atherosclerosis) (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE9874). They used baseline macrophages, monocyte-derived macrophages from peripheral blood, and foam cells cultured with or without oxidised LDL. Fifteen individuals with subclinical atherosclerosis and a family history of coronary heart disease (CHD) provided the macrophages. 15-year-olds’ macrophages and sex-matched individuals without atherosclerosis or a family history of CHD served as controls. Four lists of differentially expressed genes (DEGs) were obtained by comparing the atherosclerosis samples with normal samples of the corresponding samples for the purpose of differential gene expression analysis[30-39].
In conclusion, processing raw files, calculating intensity, and normalisation are the fundamental procedures for the majority of the study. The most popular methods for normalisation are EB, RMA, and GCRMA. Here, we have normalised raw intensity using EB. Following normalisation, we move forward with our objective, which is to comprehend the patterns of gene expression and their deduced roles. The built-in function GEO2R has been utilised for statistical analysis and differential gene expression prediction. In this instance, a matrix of gene expression levels is required, where each row corresponds to a gene and each column to a replication. The number of rows in both the normal and target data categories must be equal, and each class’s variances must be equal. We created our own algorithm for pathway and network analysis and conducted pathway research using the KEGG database.
Throughout the work, FunCoup2.0 was utilised to generate the DEGs networks, and Cytoscape was utilised to visualise the networks. We have used MATLAB for the majority of our coding and computations. Four sorts of functional couplings or linkages, including protein complexes, physical interactions between proteins, metabolic processes, and signalling pathways, are predicted by FunCoup.
Docking approach details:
ACVR1B, COL1A, CR2, GP5, IFNA21, IGF1, IL5RA, IL20RA, MAP2K6, RAPGEF3, RRM2; and BCL3, BMPRB1, CCL4, CCL5, CD14, CIITA, CSF2RA, CXCL5, HLA-DRB4, IL7R, IL15RA protein sequences were obtained from the UniProt database (www.uniprot.org). PubChem was used to obtain the 3D structures of marine drugs i.e., Alginate-n (Pubchem CID: 91666318) and Fucoidan (Pubchem CID: 129532628) in SDF format. The structure of proteins and ligands was visualized using PyMol.
The Swiss Model website (www.swissmodel.org) was used to model the homology of the proteins listed above. The GMQE, QMEANDisCo, and QMEAN Z-score analyses were used to choose the simulated structures. A scale of 0 to 1 for overall model quality is provided by GMQE (Global Model Quality Estimate) and QMEANDisCo global, where larger numbers denote higher expected quality. With all options set to default, the Swiss PDB Viewer was used to minimise the energy of the proteins’ three-dimensional structure after hydrogen atoms were introduced[38, 40].
The total number of active sites, together with information on their amino acid sequence, cavity locations, and cavity average volume, were found using a search tool. Therefore, using the Discovery Studio and CASTp server with the default probe radius (1.4 Ã…), the binding pocket of each of the proteins listed above was predicted.
PyRx was used for the molecular docking experiment (AutoDock Vina). For docking, the atomic coordinates of the protein and ligand were converted to pdbqt files. AutoDock Vina and grid box dimensions with specified spacing and size pointing in the x, y, and z directions were used to build the binding pocket[38, 39, 41-47]. The docking trials were conducted using the default parameters. To identify the best binding configurations for the compounds, the lowest binding free energy (delta G), the quantity of hydrogen bonds, and other hydrophobic interactions were also taken into consideration. Discovery Studio and PyMol examined a wide range of interactions, including hydrogen bonds, carbon-hydrogen bonds, van der Waals interactions, pi-sigma, pi-sulfur, alkyl, pi-alkyl, pi-pi T-shaped, and halogen connections.
- Results
3.1. Comparative gene expression profiling: We looked at the total gene expression profile as well as the important pathways that are enriched at a very high fold change threshold, not only the atherosclerotic genes. Genes that are upregulated, genes that are downregulated, and pathways are shown as nodes that are connected by strings (Figure 1a-1c). Here are the patterns of top upregulated genes’ interactions with other overexpressed genes and functional pathways. Upregulated genes that interact with important pathways include COL1A2, MAP2K6, RRM2, IL20RA, RAPGEF3, ITGB1, GP5, ACVR1B, CR2, IL5RA, IFNA21, IGF1, EGF, and others. In the meantime, EGF has been discovered to interact directly with FoxO signaling, cytokine-cytokine receptor interaction, ErbB signaling, and HIF-1 signaling pathways, as well as indirectly with complement and coagulation cascades, hematopoietic cell lineage, and B-cell receptor signaling pathways through upregulated CR2. Through MAP2K6, IFNA21 is linked to neuroactive ligand receptor interaction, toll-like receptor signaling, cytokine-cytokine receptor interaction pathways, and TNF signaling (Figure 1a—1c).
| Figure 1. Atherosclerosis gene expression profiling, functional analysis, and network level understanding. (a) DEGs and the inferred pathways network. (b) Up-regulated genes and (c) down-regulated genes and inferred pathways network. (d) Pathways enriched for up-regulated genes and (e) the pathways enriched for down-regulated genes. |
3.2. Functional profiling: We evaluated the impact of gene expression changes at the functional level using pathway enrichment analysis after creating the list of differentially expressed genes. We acquire a list of routes with their p-values from pathway enrichment analysis, which we utilize to determine statistical significance. Following the compilation of a list of differentially expressed genes, we used pathway enrichment analysis to assess the functional significance of gene expression variations. Pathway enrichment analysis provided us with a list of pathways and their p-values, which we used to establish statistical significance. The top 13 and 25 DEGs down and upregulated genes that were significantly modified in atherosclerosis were found using the Enriched KEGG pathways, as shown in Figure 1d—1e. The cytokine-cytokine receptor interaction signaling, PI3K—Akt signaling pathway.
3.3. Critical infectious and inflammatory pathways are dominantly affected as a result of atherosclerosis progression: To analyze the detailed role of the altered genes in terms of expression at functional level, we have performed the individual pathway level analysis followed by their pathway components. Here, we observed the significantly altered pathways and majority of the genes belong to cytokine, TLR, NF–kB, ubiquitin proteasomal system, and some more pathways also as potential components of immune system (Figure 1). Furthermore, we have also performed gene ontology (GO) terms enrichment analysis and protein classification by using panther database as shown in Figure 2. In molecular functions, binding and catalytic activity appear dominantly enriched followed by molecular transducer activity, molecular function regulator, transcription regulator activity, and transporter activity. In cellular components terms, cellular anatomical entity and protein-containing complex terms were enriched. The potential biological processes terms which were enriched potentially are: cellular process, biological regulation, metabolic process, response to stimulus, signaling, localization, multicellular organismal process, localization, and immune system process. In panther protein classification, metabolic interconversion enzyme, protein modifying enzyme, gene-specific transcriptional regulator, defense/immunity protein, protein modifying enzyme, transporter, transmembrane signal receptor, scaffold/adaptor protein, and protein-binding activity modulator (Figure 2).
| Figure 2. Functional analysis and protein classification by using Panther database. Molecular functions, cellular components, biological processes, and panther protein classes. |
3.4. Docking profiling reveals potential putative targets against atherosclerosis: Finally, we looked at herbal medicines as prospective treatments for such globally prevalent human disorders. This research not only provides prospective herbal medications, but also possible indicators.
3.4.1. Modelled structure of proteins: Supplementary Table 1 lists the modelled structures of BCL3, BMPRB1, CCL4, CCL5, CD14, CIITA, CSF2RA, CXCL5, HLA-DRB4, IL7R, IL15RA: and ACVR1B, COL1A, CR2, GP5, IFNA21, IGF1, IL5RA, IL20RA, MAP2K6, RAPGEF3, RRM2 based on GMQE and QME The fraction of residues present in most favorable areas, extra allowed regions, generously allowed regions, and banned regions are depicted by the Ramachandran plot (Suppl. Figure 1 & Supplementary Table 1). These results revealed that the modelled structures were of sufficient quality to be used in subsequent molecular docking research.
3.4.2. Binding patterns of proteins with marine drugs: Molecular docking findings revealed that the marine medicines Alginate-n and Fucoidan bind to the proteins’ expected binding site (Figures 3 and 4). Table 1 shows the minimal binding energies of proteins with marine pharmaceuticals in kcal/mol. The majority of proteins have a strong affinity for Fucoidan (Figure 3). The maximum binding affinity was found in BMPRB1, CSF2RA, MAP2K6, CIITA, and ACVR1B, with binding energies of -6.5kcal/mol, -6.3kcal/mol, -6.2kcal/mol, and -6.0kcal/mol, respectively. Except for CIITA, which displayed a binding affinity for Alginate-n, they all showed affinity for Fucoidan. We looked into the generated complexes with Discovery studio and PyMol to discover what was causing the differences in binding energies (Figure.). In terms of traditional hydrogen bonds, CSF2RA, MAP2K6, CIITA, and ACVR1B bound to Alginate-n and Fucoidan in the following order: 4, 5, 7, 8, and 2. (Table 1 and Figure 3 and 4). BMPRB1 formed substantial hydrogen connections with Tyr248, Asp350, and Lys336 with bond lengths ranging from 2.16–3.07nm. With Arg43, Thr44, Met45, and Arg155, CSF2RA demonstrated typical hydrogen bonds with bond lengths ranging from 2.11–2.69Ã…. Gly65, Lys82, Lys181, Ser201, and Lys210 formed conventional hydrogen bonds with Gly65, Lys82, Lys181, Ser201, and Lys210, with bond lengths ranging from 2.03Ã…–2.89Ã… (Figure 3 and 4). With Glu565, Ser567, Arg839, Thr866, and Trp896, Câ…¡TA formed conventional hydrogen bonds with lengths ranging from 2.14 Ã… to 2.77 Ã…. ACVR1B established conventional hydrogen bonds with Lys234 and Ser282 with bond lengths 2.18Ã… to 2.37Ã…, respectively (Figure 3 and 4). Furthermore, the respective Rama
| Figure 3. Docking profiling of up-regulated genes (inferred proteins). It represents the respective protein structures with the binding sites as enlarged view. |
chandran plots have been presented in supplementary Figure 1.
Table 1: List of top enzyme-ligand complex showing remarkable binding energies (Up and down regulated genes’ respective proteins).
|
S. No. |
Enzyme-ligand complex |
Binding energy (Kcal/mol) |
Number of HBs |
|
1. |
ACVR1B_Fucoidan |
-6.0 |
2 |
|
2. |
COL1A_Fucoidan |
-5.4 |
3 |
|
3. |
CR2_Fucoidan |
-4.7 |
3 |
|
4. |
GP5_Fucoidan |
-5.1 |
4 |
|
5. |
IFNA21_Fucoidan |
-5.9 |
3 |
|
6. |
IGF1_Fucoidan |
-4.9 |
6 |
|
7. |
IL5RA_Fucoidan |
-5.6 |
6 |
|
8. |
IL20RA_Fucoidan |
-5.1 |
4 |
|
9. |
MAP2K6_Fucoidan |
-6.3 |
7 |
|
10. |
RAPGEF3_Fucoidan |
-5.8 |
6 |
|
11. |
RRM2_Fucoidan |
-5.9 |
3 |
| Down regulated genes | |||
|
1. |
BCL3_Fucoidan |
-5.1 |
5 |
|
2. |
BMPRB1_Fucoidan |
-6.5 |
4 |
|
3. |
CCL4_Fucoidan |
-5.1 |
3 |
|
4. |
CCL5_Fucoidan |
-4.3 |
5 |
|
5. |
CD14_Alginate-n |
-5.0 |
4 |
|
6. |
Câ…¡TA_Alginate-n |
-6.2 |
7 |
|
7. |
CSF2RA_Fucoidan |
-6.5 |
4 |
|
8. |
CXCL5_Fucoidan |
-4.3 |
3 |
|
9. |
HLA-DRB4_Fucoidan |
-4.6 |
5 |
|
10. |
IL7R_Fucoidan |
-4.8 |
6 |
|
11. |
IL15RA_Alginate-n |
-4.8 |
4 |
Â
- Discussion
In order to identify potential targets and herbal remedies that can interfere with the signalling pathways, we proposed in this study to examine signalling pathways, especially inflammatory pathways, and their constituents in individual atherosclerosis patients and subgroups. In particular, we identify the genes that are up- or down-regulated in each patient with atherosclerosis using a machine learning technique, and then we search for likely activated transcription factors. After dividing atherosclerosis patients and controls into groups, we were able to identify the upstream/downstream signalling pathways of inflammatory signalling pathways. Lastly, we determined the medicines’ upstream and downstream signalling targets for alginate-N and fucoidan.
Multifocal, smouldering, immunoinflammatory atherosclerosis affects both big and medium-sized arteries and is driven by lipids. Leukocytes, intimal smooth muscle cells, and endothelial cells are the main contributors to the development of this illness. Superimposed thrombosis is responsible for the most severe effects of atherosclerosis, including heart attacks and strokes. Therefore, the key question is not why atherosclerosis occurs but rather why it abruptly becomes complex with luminal thrombosis after years of indolent progression. Atherosclerosis would be a far less dangerous condition if thrombosis-prone plaques could be identified and prevented. Plaque rupture is the cause of about 76% of all fatal coronary thrombi. Approximately 80% of coronary thrombosis in men is caused by plaque rupture, compared to 60% in women. Large lipid-rich core, thin fibrous cap with many macrophages and few smooth muscle cells, angiogenesis, adventitial inflammation, and outward remodelling are the hallmarks of ruptured plaques. The most frequent cause of coronary thrombosis is plaque rupture. The pathoanatomical characteristics of ruptured plaques and, thus, rupture-prone plaques may be helpful for imaging-based in vivo detection[1, 3, 4, 7, 13, 14].
The primary cause of cardiovascular disease, a major global cause of death, is atherosclerosis, a chronic inflammatory condition of the blood vessels. Macrophages, T lymphocytes, and other immune response cells are present in atherosclerotic lesions together with blood-borne cholesterol. In mouse models, disease is accelerated by interfering with regulatory immunity, while it is reduced by targeted deletion of genes encoding costimulatory factors and proinflammatory cytokines. Both innate and adaptive immunological responses have been linked to atherosclerosis; along the course of the illness, components of low-density lipoprotein that carry cholesterol cause inflammation, T cell activation, and antibody formation. Normal arterial defence relies on endothelial cells mounting innate immune responses and, following an inflammatory challenge, on macrophages and other immune response cells attracted to the artery wall. The development of atherosclerosis is also significantly influenced by these innate immune responses. Both internalising and signalling pattern-recognition receptors are involved[7, 9, 11, 48-52].
Numerous processes, including LDL oxidation, endothelial cell dysfunction, lipoprotein level variation, molecule adhesion, SMC migration, plaque formation, and others, are part of the intricate pathophysiology of atherosclerosis. The therapeutic medications used to treat atherosclerosis function in one or more ways. Plants have a wide range of chemical components, and these components work in different ways. Although phytochemicals and herbal remedies are a fantastic way to combat the risk of atherosclerosis, their application in the disease’s therapy is still quite limited. Traditional medicine is currently receiving attention due to the research and development of several herbal remedies for the treatment of atherosclerosis. Additionally, we found that medicinal compounds produced from herbal sources offer a larger benefit due to less side effects when compared to synthetic drugs in terms of risk and benefit ratios. Depending on how they work, using herbal medicines sparingly can provide an alternate platform for the treatment of atherosclerosis[5, 21, 53-55].
Based on the previous scientific research, this study may have concentrated on phytomedicines that have the ability to affect the cardiovascular system, specifically atherosclerosis, in terms of their safety and effectiveness. Conversely, the majority of phytomedicines have a range of cardiovascular effects that frequently overlap. This classification is not intended to assign herbs to particular illnesses, but rather to make things simpler. The dilution of active ingredients in herbal medicines has less harmful and unwanted effects than the concentration of active ingredients in allopathic treatments. Cardiovascular disease is a serious health risk, thus it is important to pay attention to these side effects and drug interactions. No herbal medicine program should be initiated without carefully assessing the possible outcomes.
5. Conclusions
The goal of this research is to identify potential targets and herbal remedies that may interfere with the active signaling pathways that lead to individual atherosclerosis. In particular, we have identified the genes that are up- or down-regulated in particular atherosclerosis patients using a machine learning technique, and then we search for likely activated transcription factors. We identified several subgroups of atherosclerosis patients with similar transcription factors based on their upstream/downstream signaling pathways. Lastly, we determined which upstream and downstream signaling targets the medications alginate-n and fucoidan target.
Supplementary Materials: The following supporting information can be downloaded at: www.jbsciences.com/xxx/jbs-2024006-SupplementaryFigure1.pptx (Figure S1); www.jbsciences.com/jbs-2024006-Supplementary_Table.docx (Table S1).
Author Contributions: Conceptualization, S.A., R.J., and M.M.; methodology, S.A., R.J., and M.M.; software, S.A., R.J., and M.M.; validation, S.A. and M.M.; formal analysis, S.A. and M.M.; investigation, S.A. and M.M.; resources, S.A. and M.M.; data curation, S.A. and M.M.; writing—original draft preparation, S.A., R.J., and M.M.; writing—review and editing, S.A. and M.M.; visualization, S.A. and M.M.; supervision, S.A. and M.M.; project administration, S.A. and M.M.; funding acquisition, S.A. and M.M. All authors have read and agreed to the published version of the manuscript.
Funding: Not applicable.
Acknowledgments: We are grateful to the Department of Biochemistry, Faculty of Sciences, University of Tabuk, Tabuk, 71491, Saudi Arabia, Department of Biomedical Laboratory Science, NTNU, NO-7491 Trondheim, Norway, and G D College, Begusarai, 851101, Bihar, India for providing us all the facilities to carry out the entire work.
Conflicts of Interest: The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Institutional Review Board Statement: Not applicable.
Informed Consent Statement: Not applicable.
Data Availability Statement: All the related data are supplied in this work or have been referenced properly.
References
- Beckman, R. & Loeb, L. Genetic instability in cancer: Theory and experiment. Seminars in Cancer Biology 15,423–435 (2005).
- Joenje, H. & Patel, K. J. The emerging genetic and molecular basis of Fanconi anaemia. Nature Reviews Genetics 2, 446–457 (2001).
- Swanton, C. Cancer evolution: the final frontier of precision medicine? Annals of Oncology 25, 549–551 (2014).
- Michor, F., Iwasa, Y. & Nowak, M. A. Dynamics of cancer progression. Nature Reviews Cancer 4, 197–205 (2004).
- Paguirigan, A. L. et al. Single-cell genotyping demonstrates complex clonal diversity in acute myeloid leukemia. Science Translational Medicine 7, (2015).
- Yizhak, K. et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science 364, eaaw0726 (2019).
- Cooper, C. S. et al. Analysis of the genetic phylogeny of multifocal prostate cancer identifies multiple independent clonal expansions in neoplastic and morphologically normal prostate tissue. Nature Genetics 1–9 (2015). doi:10.1038/ng.3221
- Wheeler, H. E., Maitland, M. L., Dolan, M. E., Cox, N. J. & Ratain, M. J. Cancer pharmacogenomics: strategies and challenges. Nature Reviews Genetics 14, 23–34 (2012).
- Pon, J. R. & Marra, M. A. Driver and Passenger Mutations in Cancer. Annu. Rev. Pathol. Mech. Dis. 10, 25–50 (2015).
- Vogelstein, B. et al. Cancer genome landscapes. Science 339, 1546–1558 (2013).
- Parsons, D. W. et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science 321, 1807–1812 (2008).
- Klijn, C. et al. Analysis of Tumor Heterogeneity and Cancer Gene Networks Using Deep Sequencing of MMTV-Induced Mouse Mammary Tumors. PLoS ONE 8, e62113 (2013).
- Huang, S., Ernberg, I. & Kauffman, S. Cancer attractors: A systems view of tumors from a gene network dynamics and developmental perspective. Seminars in Cell & Developmental Biology 20, 869–876 (2009).
- Wang, E. Cancer Letters. Cancer Letters 340, 261–269 (2013).
- Stein, S. et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nature Medicine 16, 198–204 (2010).
- Gazy, I. & Kupiec, M. Genomic instability and repair mediated by common repeated sequences. Proceedings of the National Academy of Sciences 110, 19664–19665 (2013).
- Habermann, J. K. et al. The gene expression signature of genomic instability in breast cancer is an independent predictor of clinical outcome. Int. J. Cancer 124, 1552–1564 (2009).
- Grubor, V. et al. Novel genomic alterations and clonal evolution in chronic lymphocytic leukemia revealed by representational oligonucleotide microarray analysis (ROMA). Blood 113, 1294–1303 (2009).
- Restifo, N. P., Smyth, M. J. & Snyder, A. Acquired resistance to immunotherapy and future challenges. Nature Reviews Cancer 16, 121–126 (2016).
- Zhao, B., Pritchard, J. R., Lauffenburger, D. A. & Hemann, M. T. Addressing Genetic Tumor Heterogeneity through Computationally Predictive Combination Therapy. Cancer Discovery 4, 166–174 (2014).
- Fisher, R., Pusztai, L. & Swanton, C. Cancer heterogeneity: implications for targeted therapeutics. British Journal of Cancer 108, 479–485 (2013).
- Alexander, S. & Friedl, P. Cancer invasion and resistance: interconnected processes of disease progression and therapy failure. Trends in Molecular Medicine 18, 13–26 (2012).
- Farazi, T. A. et al. Bioinformatic analysis of barcoded cDNA libraries for small RNA profiling by next-generation sequencing. Methods 58, 171–187 (2012).
- Rizzo, J. M. & Buck, M. J. Key Principles and Clinical Applications of ‘Next-Generation’ DNA Sequencing. Cancer Prevention Research 5, 887–900 (2012).
- Robasky, K., Lewis, N. E. & Church, G. M. The role of replicates for error mitigation in next-generation sequencing. Nature Reviews Genetics 15, 56–62 (2013).
- Malumbres, M. & Barbacid, M. Cell cycle, CDKs and cancer: a changing paradigm. Nature Reviews Cancer 9,153–166 (2009).
- Heenan, E. J. & Sancar, A. DNA Damage: Repair. (John Wiley & Sons, Inc., 2007). doi:10.1002/9780470048672.wecb131
- Raffoul, J. J., Heydari, A. R. & Hillman, G. G. DNA Repair and Cancer Therapy: Targeting APE1/Ref-1 Using Dietary Agents. Journal of Oncology 2012, 1–11 (2012).
- Polo, S. E. & Jackson, S. P. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes & Development 25, 409–433 (2011).
- Forment, J. V. & O’Connor, M. J. Accepted Manuscript. #startpage# (2018). doi:10.1016/j.pharmthera.2018.03.005
- Harris, R. S. Cancer mutation signatures, DNA damage mechanisms, and potential clinical implications. 5, 1–1 (2013).
- Hoadley, K. A. et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 173, 291–304.e6 (2018).
- Alexandrov, L. B. et al. Clock-like mutational processes in human somatic cells. Nature Genetics 47, 1402–1407 (2015).
- Watson, I. R., Takahashi, K., Futreal, P. A. & Chin, L. Emerging patterns of somatic mutations in cancer. Nature Reviews Genetics 14, 703–718 (2013).
- Ferreira, M. A. et al. Genome-wide association and transcriptome studies identify target genes and risk loci for breast cancer. Nature Communications 1–18 (2019). doi:10.1038/s41467-018-08053-5
- Helleday, T., Eshtad, S. & Nik-Zainal, S. Mechanisms underlying mutational signatures in human cancers. Nature Reviews Genetics 15, 585–598 (2014).
- Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
- Burrell, R. A. & Swanton, C. ScienceDirectThe evolution of the unstable cancer genome. Current Opinion in Genetics & Development 24, 61–67 (2014).
- WARBURG, O. Origin of Cancer Cells. Science 123, 309–314 (1956).
- Ross, D. T. et al. Systematic variation in gene expression patterns in human cancer cell lines. Nature Genetics24, 227–235 (2000).
- Kittler, R. et al. A Comprehensive Nuclear Receptor Network for Breast Cancer Cells. CellReports 3, 538–551 (2013).
- Ramaswamy, S., Ross, K. N., Lander, E. S. & Golub, T. R. A molecular signature of metastasis in primary solid tumors. Nature Genetics 33, 49–54 (2002).
- Pereira, B. et al. The somatic mutation profiles of 2,433 breast cancers refine their genomic and transcriptomic landscapes. Nature Communications 7, 1–15 (2016).
- Liu, J. et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 173, 400–416.e11 (2018).
- Huang, D. W., Sherman, B. T. & Lempicki, R. A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Research 37, 1–13 (2009).
- Li, Y., Vongsangnak, W., Chen, L. & Shen, B. Integrative analysis reveals disease-associatedgenes and biomarkers for prostate cancerprogression. BMC Medical Genomics 7, S3 (2014).
- Chen, Y. et al. Identifying potential cancer driver genes by genomic data integration. Sci. Rep. 3, (2013).
- Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Research 13, 2498–2504 (2003).
- Newman, R. H. et al. Construction of human activity-based phosphorylation networks. Molecular Systems Biology 9, 1–12 (2013).
- Kamburov, A., Stelzl, U., Lehrach, H. & Herwig, R. The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Research 41, D793–800 (2013).
- Anaya, J. OncoLnc: linking TCGA survival data to mRNAs, miRNAs, and lncRNAs. PeerJ Computer Science2, e67 (2016).
- Mi, H., Poudel, S., Muruganujan, A., Casagrande, J. T. & Thomas, P. D. PANTHER version 10: expanded protein families and functions, and analysis tools. Nucleic Acids Research 44, D336–42 (2016).
- Kumar, P. P. et al. In-silico study reveals immunological signaling pathways, their genes, and potential herbal drug targets in ovarian cancer. Informatics in Medicine Unlocked 100422 (2020).
- Warsi, M. K., Kamal, M. A., Baeshen, M. N., Izhari, M. A. & Mobashir, A. F. A. M. Comparative Study of Gene Expression Profiling Unravels Functions associated with Pathogenesis of Dengue Infection. Current Pharmaceutical Design 26 IS , 1–8
- Kamal, M. A. et al. Gene expression profiling and clinical relevance unravel the role hypoxia and immune signaling genes and pathways in breast cancer: Role of hypoxia and immune signaling genes in breast cancer. jimsa 1, (2020).
- Fraser, M. et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature 1–22 (2017). doi:10.1038/nature20788
- Biesecker, L. G. & Spinner, N. B. A genomic view of mosaicism and human disease. Nature Reviews Genetics14, 307–320 (2013).
- Aguilera, A. & GarcÃa-Muse, T. Causes of Genome Instability. Annu. Rev. Genet. 47, 1–32 (2013).
- Speleman, F. et al. Copy number alterations and copy number variation in cancer: close encounters of the bad kind. Cytogenet Genome Res 123, 176–182 (2009).
- Gaillard, H., GarcÃa-Muse, T. & Aguilera, A. Replication stress and cancer. Nature Reviews Cancer 15, 276–289 (2015).
- Roberts, S. A. & Gordenin, D. A. Hypermutation in human cancer genomes: footprints and mechanisms. Nature Reviews Cancer 14, 786–800 (2014).
- Helleday, T., Eshtad, S. & Nik-Zainal, S. Mechanisms underlying mutational signatures in human cancers. Nature Reviews Genetics 15, 585–598 (2014).
- Nik-Zainal, S. et al. Mutational Processes Molding the Genomes of 21 Breast Cancers. Cell 149, 979–993 (2012).
- Alexandrov, L. B., Nik-Zainal, S., Wedge, D. C., Campbell, P. J. & Stratton, M. R. Deciphering Signatures of Mutational Processes Operative in Human Cancer. CellReports 3, 246–259 (2013).
- Agell, L. et al. A 12-Gene Expression Signature Is Associated with Aggressive Histological in Prostate Cancer. The American Journal of Pathology 181, 1585–1594 (2012).
- Sebag, A. S., Tomkiewicz-Raulet, C. & Barouki, R. A generic methodological framework for studying single cell motility in high-throughput time-lapse screening data. (The Febs …, 2014). doi:10.1093/bioinformatics/btv225/-/DC1
- Stelzl, U. et al. A Human Protein-Protein Interaction Network: A Resource for Annotating the Proteome. Cell122, 957–968 (2005).
- Stoevesandt, O. et al. A Network Analysis of Changes in Molecular Interactions in Cellular Signaling. Molecular & Cellular Proteomics 6, 503–513 (2006).
- Fujiki, Y. et al. APOBEC3B gene expression as a novel predictive factor for pathological complete response to neoadjuvant chemotherapy in breast cancer. Oncotarget 9, 30513–30526 (2018).
- Mao, Y. et al. APOBEC3B expression and its prognostic potential in breast cancer. Oncol Lett (2020). doi:10.3892/ol.2020.11433
- Kim, Y.-S. et al. Clinical implications of APOBEC3A and 3B expression in patients with breast cancer. PLoS ONE 15, e0230261 (2020).
- Zhang, Y., Delahanty, R., Guo, X., Zheng, W. & Long, J. Integrative genomic analysis reveals functional diversification of APOBEC gene family in breast cancer. Human Genomics 1–12 (2015). doi:10.1186/s40246-015-0056-9
- Matsumoto, T. et al. protein kinase A inhibits tumormutator APOBEC3B throughphosphorylation. Sci. Rep. 1–12 (2019). doi:10.1038/s41598-019-44407-9
- Yamazaki, H. et al. APOBEC3B reporter myeloma cell lines identify DNA damage response pathways leading to APOBEC3B expression. PLoS ONE 15, e0223463 (2020).
- Sanchez-Vega, F. et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 173, 321–337.e10 (2018).
- Guttridge, K. H. F. D. G. D., Glass, D. J. & Guttridge, D. C. Cancer Cachexia:Mediators, Signaling, and Metabolic Pathways. Cell Metabolism 16, 153–166 (2012).
- Brechbiel, J., Miller-Moslin, K. & Adjei, A. A. Crosstalk Between Hedgehog and Other Signaling Pathways as a Basis for Combination Therapies in Cancer. Cancer Treatment Reviews 40, 750–759 (2014).
- Eroles, P., Bosch, A., Pérez-Fidalgo, J. A. & Lluch, A. Cancer Treatment Reviews. Cancer Treatment Reviews38, 698–707 (2012).
- da Silva, H. B. et al. Dissecting Major Signaling Pathways throughout the Development of Prostate Cancer. Prostate Cancer 2013, 1–23 (2013).
- Oren, M. & Aylon, Y. The Hippo Signaling Pathway and Cancer. (Springer Science & Business Media, 2013).
- Altomare, D. A. & Testa, J. R. Perturbations of the AKT signaling pathway in human cancer. Oncogene 24,7455–7464 (2005).
- Wilson, F. H. A Cluster of Metabolic Defects Caused by Mutation in a Mitochondrial tRNA. Science 306, 1190–1194 (2004).
- Bansal, M. et al. a community computational challenge to predict the activity of pairs of compounds. Nat Biotechnol 1–12 (2014). doi:10.1038/nbt.3052
- Nakamura, H. et al. Genomic spectra of biliary tract cancer. Nature Genetics 1–11 (2015). doi:10.1038/ng.3375
- Real, F. X., Boutros, P. C. & Malats, N. Next-generation Sequencing of Urologic Cancers: Next Is Now. European Urology 66, 4–7 (2014).
- Mobashir, M., Schraven, B. & Beyer, T. Simulated evolution of signal transduction networks. PLoS ONE 7,e50905 (2012).
- Mobashir, M., Madhusudhan, T., Isermann, B., Beyer, T. & Schraven, B. Negative Interactions and Feedback Regulations Are Required for Transient Cellular Response. Sci. Rep. 4,
- Abollo-Jiménez, F., Jiménez, R. & Cobaleda, C. Physiological cellular reprogramming and cancer. Seminars in Cancer Biology 20, 98–106 (2010).
- Ferrell, J. E., Jr. The Biochemical Basis of an All-or-None Cell Fate Switch in Xenopus Oocytes. Science 280,895–898 (1998).
- Chatterjee, A., Kaznessis, Y. N. & Hu, W.-S. Tweaking biological switches through a better understanding of bistability behavior. Current Opinion in Biotechnology 19, 475–481 (2008).
- Iwamoto, K., Hamada, H., Eguchi, Y. & Okamoto, M. Mathematical modeling of cell cycle regulation in response to DNA damage: Exploring mechanisms of cell-fate determination. Biosystems 103, 384–391 (2011).
- Mustafa, S. & Mobashir, M. LC–MS and docking profiling reveals potential difference between the pure and crude fucoidan metabolites. International Journal of Biological Macromolecules 143, 11–29 (2020).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of JBS and/or the editor(s). JBS and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
Copyright: © 2024 by the authors. Submitted for possible open access publication under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
![]()